
La finalización del genoma humano completo «telómero a telómero» (T2T) el año pasado enfatizó que las secuencias del genoma que anteriormente se pensaba que estaban «completas», de hecho, no estaban completas en absoluto. Crédito: El Laboratorio Jackson
La finalización del genoma humano completo «telómero a telómero» (T2T) el año pasado enfatizó que las secuencias del genoma que anteriormente se pensaba que estaban «completas», de hecho, no estaban completas en absoluto.
Además, muchos genomas recientes se secuencian con tecnologías de secuenciación de lectura corta, que fragmentan el ADN en segmentos cortos, normalmente de 150 a 300 pares de bases, y luego se comparan con una secuencia de referencia. Si bien las metodologías de lectura corta son rápidas, precisas y relativamente económicas, habitualmente pasan por alto grandes partes del genoma, alrededor del 10 % en total. Los segmentos que faltan incluyen regiones de alto contenido de G/C y secuencias repetitivas, incluidas duplicaciones segmentarias, repeticiones simples y elementos transponibles (TE).
Los TE son secuencias repetitivas que se han movido a otras ubicaciones en el genoma, y la movilidad de estas secuencias contribuye en gran medida a la variación genómica. Las secuencias repetitivas subyacen con frecuencia a la formación de variantes estructurales (SV), diferencias genómicas resultantes de duplicaciones, inserciones, eliminaciones e inversiones. Los SV a menudo se pasan por alto cuando se usa secuenciación de lectura corta (en particular, aquellos mediados por repeticiones), pero pueden desempeñar un papel importante en la desregulación del genoma y la enfermedad.
Los investigadores han recurrido a la secuenciación de lectura larga para analizar los genomas de manera más completa, ya que estas tecnologías permiten la secuenciación de segmentos de ADN mucho más largos y pueden capturar con precisión una imagen más completa de un genoma. Los avances recientes han mejorado la precisión y la utilidad de la lectura larga, lo que permite a los investigadores investigar características genómicas no detectadas anteriormente, y no solo en humanos.
Jackson Laboratory (JAX) y la profesora asistente del Centro de Salud de la Universidad de Connecticut, Christine Beck, Ph.D., dirigieron un equipo que exploró los genomas de otra especie notable, el ratón, y reveló detalles en 20 cepas endogámicas diversas que serán fundamentales para informar Avanza la investigación en genética y genómica basada en ratones.
Variación estructural entre cepas de ratón.
Los ratones tienen su propio genoma de referencia, conocido como GRCm39, basado en la secuencia de C57BL/6J, una cepa de la subespecie Mus musculus domesticus. Pero muchas cepas de ratones de laboratorio de uso común también se han derivado de otras dos subespecies, Mus musculus castaneus y Mus musculus musculus, y existen muchas diferencias genéticas entre las diferentes cepas endogámicas.
Por el trabajo presentado en «La resolución de la variación estructural en diversos genomas de ratón revela la remodelación de la cromatina debido a elementos transponibles», publicado en Genómica Celularel Dr. Beck seleccionó una amplia variedad de cepas de uso común, incluidos los siete padres fundadores de los paneles de ratones genéticamente diversos Collaborative Cross (CC) y Diversity Outbred (DO), seis cepas CC resultantes con anomalías de origen genético desconocido y otras siete cepas de uso común con diferentes antecedentes genéticos.
Ardian Ferraj, estudiante de posgrado y autor principal del estudio, luego reunió los genomas de estos 20 ratones y usó estas secuencias para identificar los SV presentes en los animales que diferenciaban sus genomas de los de la referencia C57BL/6J. Usando PAV, un programa desarrollado por el Dr. Peter Audano, miembro del laboratorio de Beck, Ardian demostró que los SV prevalecen en los genomas de ratón y contribuyen ampliamente a la variación genómica. De hecho, los SV contienen casi cinco veces el número de bases afectadas en comparación con las variantes de nucleótido único publicadas anteriormente de diversos genomas de ratón.
También encontraron una diversidad mucho mayor de SV entre genomas de ratón que entre genomas humanos, lo que sugiere que un solo genoma de referencia de ratón es inadecuado para mapear datos genómicos entre cepas de ratones. Es importante destacar que la secuenciación de lectura larga es vital para capturar esta variación. En 18 de las cepas de ratones, el equipo de investigación detectó 213 688 inserciones adicionales, 64 277 eliminaciones y 97 inversiones con lecturas largas en comparación con datos de lectura corta.
. DOI: 10.1016/j.xgen.2023.100291")
Crédito: Genómica Celular (2023). DOI: 10.1016/j.xgen.2023.100291
Elementos transponibles y consecuencias de la variación estructural
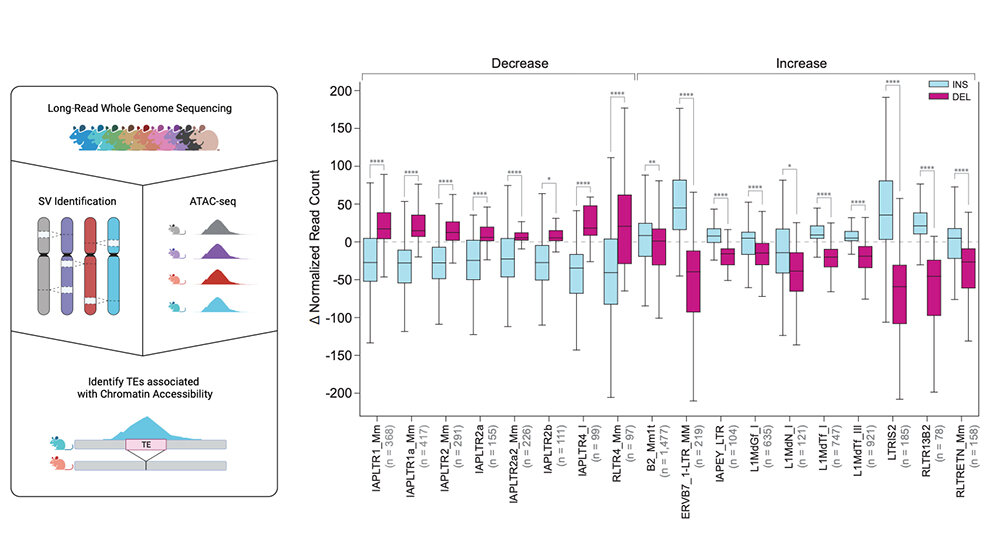
Si bien solo una pequeña cantidad de TE aún pueden movilizarse en los genomas humanos, son más móviles en ratones. Debido a esto, Beck y su equipo se centraron en las variantes de elementos transponibles (TEV), que descubrieron que comprendían casi el 40 % de todos los SV, y la mayoría (60 %) eran inserciones. Hay varios tipos de TEV, conocidos como elementos nucleares intercalados cortos versus largos (SINE y LINE), que se caracterizan de manera predecible por su tamaño. Los LINE fueron casi el doble de comunes que los SINE en los genomas de ratón, del 47 % al 24 %.
Debido a su tamaño, los LINE también contribuyen con casi la mitad del contenido de secuencia variable en los genomas de ratón, en comparación con solo el 24 % que aportan los SV que no son TEV y el 2,1 % de los SINE. Varias secuencias retrovirales endógenas generaron el 28% restante de los VET. Los retrovirus son virus de ARN cuyos genomas se transcriben inversamente a ADN, que luego se inserta en el genoma. Si bien muchos retrovirus actuales están asociados con enfermedades como el SIDA y el cáncer, los genomas de mamíferos normales contienen grandes cantidades de ADN derivado de retrovirus durante milenios, conocidos como retrovirus endógenos o ERV, que ayudan a impulsar la variación genómica en ratones.
Entonces, ¿cuáles son las posibles consecuencias de toda esta variación y actividad genómica? Los investigadores observaron las SV en el contexto de las características genómicas conocidas y predijeron la gravedad de los efectos. Entre los SV recién detectados dentro de las secuencias de genes, la gran mayoría (94.863) estaban dentro de los intrones, las secuencias que se separan de los pre-ARNm para que no alteren la estructura de la proteína; 1.469 estaban en los segmentos no traducidos (UTR) en cualquier extremo del gen; y 510 dentro de las secuencias de codificación de proteínas reales.
También identificaron una inserción de elemento retroviral previamente no detectada dentro de un gen específico, Mutyh, un gen de reparación de ADN asociado con una firma mutacional conocida en ciertas cepas de ratones. Se desconocía la variante subyacente, pero el equipo encontró que la inserción estaba asociada con una disminución significativa en la expresión del gen Mutyh. El hallazgo muestra que los SV desconocidos pueden alterar regiones genómicas importantes y residir en genes asociados con rasgos relevantes para la salud y la función, incluida la enfermedad.
Finalmente, en colaboración con la investigadora de Jax, la Dra. Laura Reinholdt, el equipo investigó el impacto de los TE en las diferencias de células madre embrionarias. Los TE promueven la diversidad del genoma y su variación puede alterar aspectos importantes de la expresión génica entre cepas. De hecho, el estudio encontró más de 22 000 TEV asociados con cambios significativos en la accesibilidad a la cromatina de las células madre, un regulador clave de la expresión génica, en células madre embrionarias de 10 cepas de ratones genéticamente diversas.
Centrándose nuevamente en un ejemplo específico, investigaron una inserción intrónica específica de cepa (CAST/EiJ) en el gen Slc47a2, que iba acompañada de una señal de accesibilidad a la cromatina exclusiva de la cepa. Encontraron niveles elevados de expresión de Slc47a2 en comparación con las cepas que carecen de la inserción, con una transcripción específica de la cepa y una posible región de unión para un factor de pluripotencia, lo que indica funciones importantes para los TEV en el desarrollo temprano.
Una comprensión más completa
Dada la importancia del ratón como modelo para la genética de los mamíferos y las enfermedades humanas, es necesario comprender completamente las consecuencias funcionales de la variación genómica. La detección y caracterización integrales de SV entre genomas de cepas de ratones es una parte crucial de dicha comprensión, y los resultados y los datos generados por la Dra. Beck y sus colaboradores brindan un importante paso adelante en este campo.
Los autores produjeron un recurso SV resuelto por secuencia, un recurso de expresión de células madre embrionarias de ratón y datos de accesibilidad de la cromatina para la comunidad de investigación que pueden ayudar a futuras investigaciones sobre la evolución del ratón y la genómica subyacente a los rasgos de interés.
Más información:
Christine R. Beck, La resolución de la variación estructural en diversos genomas de ratón revela la remodelación de la cromatina debido a elementos transponibles, Genómica Celular (2023). DOI: 10.1016/j.xgen.2023.100291. www.cell.com/cell-genomics/ful … 2666-979X(23)00057-5
Citación: Un nuevo estudio revela detalles de 20 cepas de ratones consanguíneas diversas (5 de abril de 2023) consultado el 5 de abril de 2023 en https://phys.org/news/2023-04-reveals-diverse-inbred-mouse-strains.html
Este documento está sujeto a derechos de autor. Aparte de cualquier trato justo con fines de estudio o investigación privados, ninguna parte puede reproducirse sin el permiso por escrito. El contenido se proporciona únicamente con fines informativos.


