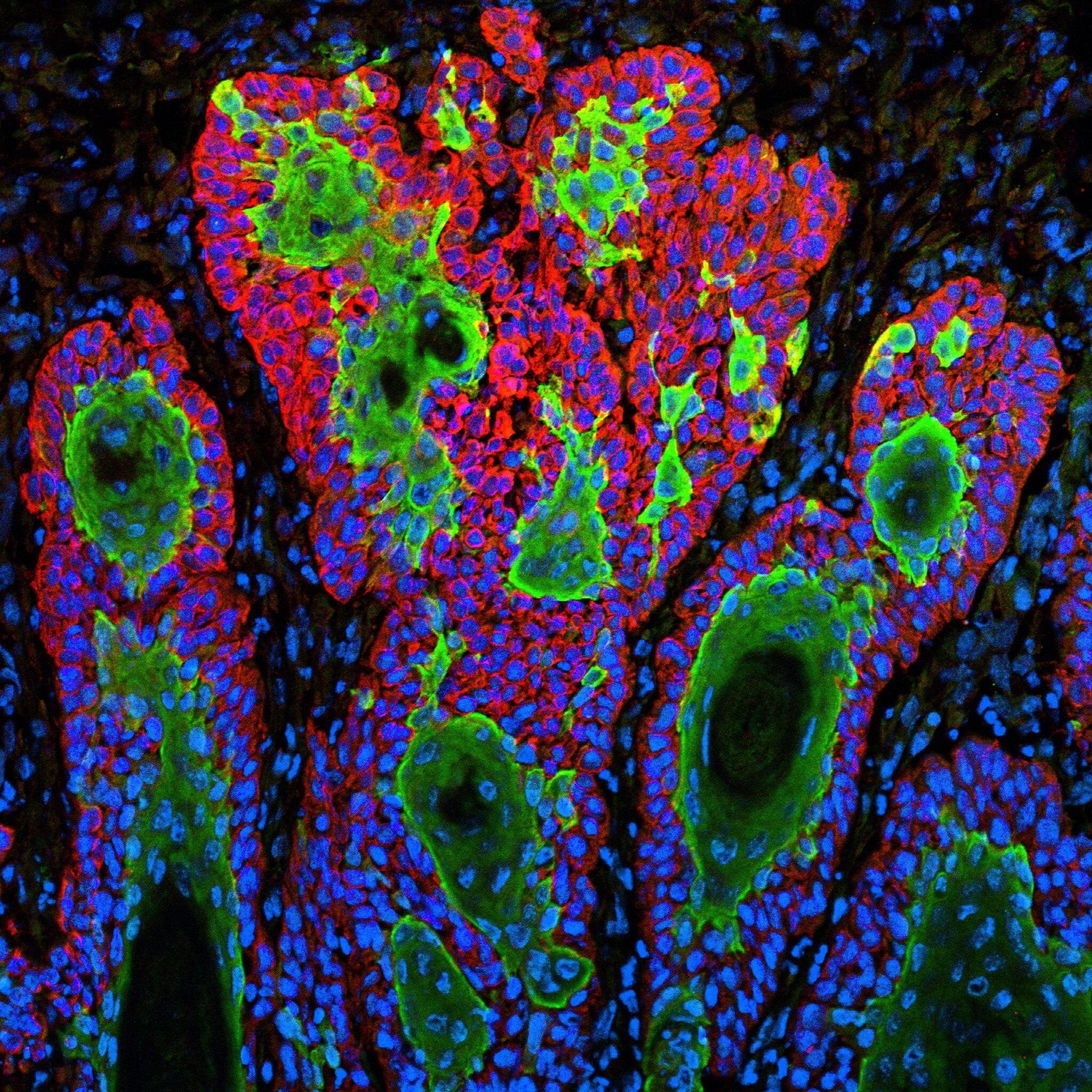
Un nuevo método desarrollado por investigadores de Princeton integra información de expresión génica de múltiples cortes tomados de la misma muestra de tejido, proporcionando una vista tridimensional de las actividades celulares en la salud y la enfermedad, incluido el carcinoma de células escamosas del cáncer de piel común. Crédito: Markus Schober y Elaine Fuchs, Universidad Rockefeller
Así como es difícil entender una conversación sin conocer su contexto, puede ser difícil para los biólogos comprender el significado de la expresión génica sin conocer el entorno de una célula. Para resolver ese problema, los investigadores de Princeton Engineering han desarrollado un método para dilucidar el entorno de una célula para que los biólogos puedan dar más significado a la información de expresión génica.
Los investigadores, dirigidos por el profesor de Ciencias de la Computación Ben Raphael, esperan que el nuevo sistema abra la puerta para identificar tipos de células raras y elegir opciones de tratamiento del cáncer con nueva precisión. Raphael es el autor principal de un artículo que describe el método publicado el 16 de mayo en Métodos de la naturaleza.
La técnica básica de vincular la expresión génica con el entorno de una célula, llamada transcriptómica espacial (ST), existe desde hace varios años. Los científicos descomponen las muestras de tejido en una cuadrícula a microescala y vinculan cada punto de la cuadrícula con información sobre la expresión génica. El problema es que las herramientas computacionales actuales solo pueden analizar patrones espaciales de expresión génica en dos dimensiones. Los experimentos que utilizan múltiples cortes de una sola muestra de tejido, como una región de un cerebro, corazón o tumor, son difíciles de sintetizar en una imagen completa de los tipos de células en el tejido.
El método de los investigadores de Princeton, llamado PASTE (Probabilistic Alignment of ST Experiments), integra información de múltiples cortes tomados de la misma muestra de tejido, proporcionando una vista tridimensional de la expresión génica dentro de un tumor o un órgano en desarrollo. Cuando la cobertura de secuencias en un experimento es limitada debido a problemas técnicos o de costo, PASTE también puede fusionar información de múltiples cortes de tejido en un solo corte de consenso bidimensional con información de expresión génica más rica.
«Nuestro método fue motivado por la observación de que, a menudo, los biólogos realizan múltiples experimentos con el mismo tejido», dijo Raphael. «Ahora, estos experimentos replicados no son exactamente las mismas células, pero son del mismo tejido y, por lo tanto, deberían ser muy similares».
La técnica del equipo puede alinear múltiples cortes de una sola muestra de tejido, clasificando las células en función de sus perfiles de expresión génica y preservando la ubicación física de las células dentro del tejido.
El proyecto comenzó en el verano de 2020 después de que Max Land, un concentrador de matemáticas de la Clase de 2021 de Princeton, tomó el curso de Raphael «Algoritmos en Biología Computacional». Emocionado por el campo en rápida evolución y la oportunidad de mejorar la comprensión de la salud y las enfermedades humanas, Land se acercó a Raphael para involucrarse en la investigación y comenzó a trabajar en el código para desarrollar lo que se convirtió en el método PASTE. Fue asesorado por Raphael y por el autor principal del estudio, Ron Zeira, un ex investigador postdoctoral en Princeton que ahora es científico investigador en la compañía de salud de precisión Verily.
El trabajo fue el foco de la tesis de graduación de Land, y él coescribió el artículo junto con Zeira, Raphael y Alexander Strzalkowski, un Ph.D. en ciencias de la computación. alumno. Ahora, un biólogo computacional en el Centro de Cáncer Memorial Sloan Kettering en la ciudad de Nueva York, Land dijo que la tutoría de Zeira y Raphael ha sido fundamental en su búsqueda de una carrera de investigación.
El equipo desarrolló su método utilizando datos de expresión génica simulada de un estudio de transcriptómica espacial de un tumor de mama, donde se estableció previamente la correspondencia entre cortes de tejido. Luego evaluaron el método con datos recopilados de muestras de la corteza prefrontal del cerebro, que tiene una estructura conocida que consiste en capas de diferentes tipos de células con firmas de expresión génica únicas.
Los investigadores también aplicaron PASTE a los datos recopilados de las biopsias de cáncer de piel de cuatro pacientes diferentes. Un análisis previo de estos datos había sugerido un mosaico complejo de tipos de células, con un alto grado de células sanas y cancerosas entremezcladas. El método PASTE, sin embargo, reveló que la aparente baja coherencia espacial en tres de las muestras de los pacientes probablemente se debió a la baja cobertura de secuencia en los experimentos. El nuevo análisis mostró que las células se agruparon en grupos más contiguos, un escenario biológicamente más plausible.
«Después de integrar varios de estos cortes y aumentar efectivamente la cobertura de los datos, obtenemos agrupaciones de células más espacialmente coherentes, lo cual es más razonable que cada tipo de célula colocado al azar en el tejido», dijo Zeira.
Hasta ahora, el conjunto de datos más grande que ha analizado el equipo fue una muestra de tejido cardíaco con nueve cortes, pero tienen la vista puesta en experimentos con embriones de ratón que incluyen más de 30 cortes. Además de las consideraciones computacionales, los experimentos de transcriptómica espacial a esta escala siguen siendo costosos para muchos laboratorios, dijo Raphael.
Aún así, agregó, «esperamos que tener una herramienta como PASTE anime a más investigadores a realizar experimentos replicados, porque ahora pueden usar la información de cortes adicionales de una manera que antes no podían hacer fácilmente».
El enfoque computacional permite el mapeo espacial de datos de una sola célula dentro de los tejidos
Ron Zeira et al, Alineación e integración de datos de transcriptómica espacial, Métodos de la naturaleza (2022). DOI: 10.1038/s41592-022-01459-6
Citación: El nuevo método combina datos para hacer un mapa tridimensional de las actividades de las células (16 de mayo de 2022) recuperado el 17 de mayo de 2022 de https://phys.org/news/2022-05-method-melds-d-cells.html
Este documento está sujeto a derechos de autor. Aparte de cualquier trato justo con fines de estudio o investigación privados, ninguna parte puede reproducirse sin el permiso por escrito. El contenido se proporciona únicamente con fines informativos.

