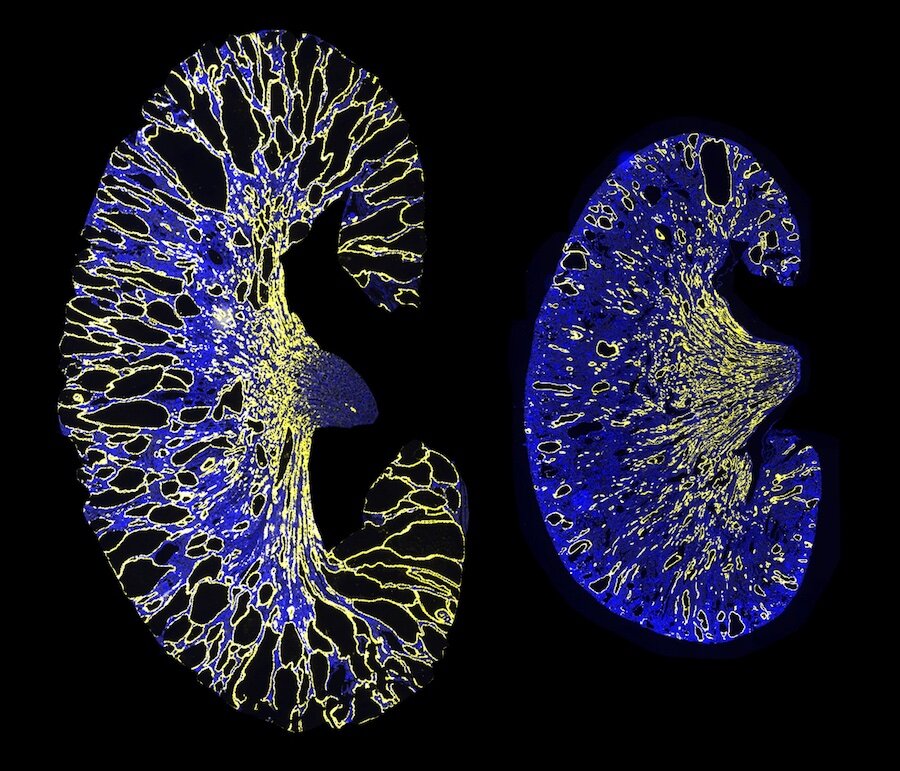
Un riñón severamente quístico de un modelo de ratón ADPKD (izquierda). La eliminación del sitio de unión de PKD1 miR-17 reduce notablemente el crecimiento de quistes (derecha). El amarillo indica los conductos colectores del riñón y el azul indica los núcleos. Crédito: UT Southwestern Medical Center
El bloqueo de la inhibición de la expresión génica de PKD1 y PKD2 mediante la eliminación de un sitio de unión para microARN obstaculizó la formación y el crecimiento de quistes renales en modelos de enfermedad renal poliquística autosómica dominante (ADPKD), informaron investigadores de UT Southwestern. Los hallazgos, publicados en Comunicaciones de la naturalezasugieren una estrategia para la terapia génica con el potencial de detener o curar la ADPKD.
«Durante más de 25 años, hemos sabido que la ADPKD es causada por mutaciones de los genes PKD1 o PKD2. Sin embargo, no existe una estrategia terapéutica para abordar estas causas fundamentales», dijo Vishal Patel, MD, Profesor Asociado de Medicina Interna en la División. de Nefrología en UTSW y autor correspondiente del artículo.
La ADPKD se encuentra entre las afecciones genéticas humanas más comunes y la causa genética más frecuente de insuficiencia renal, y afecta a aproximadamente 12,5 millones de personas en todo el mundo. ADPKD es una enfermedad hereditaria en la que los pacientes suelen heredar una copia mutada de PKD1 (o PKD2) y una copia normal.
La enfermedad se caracteriza por la formación frecuente de muchos pequeños sacos llenos de líquido llamados quistes renales, que se cree que se forman cuando los niveles de PKD1 o PKD2 caen por debajo de un umbral crítico. Esto puede ocurrir cuando la copia normal del gen no produce suficientes proteínas policistina-1/policistina-2.
Las proteínas se producen (o traducen) a partir del ácido ribonucleico mensajero (ARNm) de un gen. En un extremo de la cadena de ARNm hay una región de código que ayuda a protegerla de la degradación, pero también puede controlar la cantidad de proteína que se produce. La unión de los microARN a esta región del código del ARNm puede bloquear la traducción, lo que conduce a la producción de menos proteína.
PKD1 contiene un sitio de unión para miR-17, un microARN altamente expresado y activo en modelos de ADPKD. Entonces, el Dr. Patel y sus colegas preguntaron si el bloqueo de la unión de miR-17 a PKD1 podría prevenir la formación de quistes renales.
Los investigadores eliminaron el sitio de unión de miR-17 del ARNm de PKD1 en cultivos celulares y en un modelo de ratón con ADPKD. Sus resultados indicaron que la eliminación del sitio de unión aumentó la estabilidad de la cadena de ARNm, elevó los niveles de policistina-1 y disminuyó el crecimiento de quistes renales. Además, el grupo descubrió que bloquear la unión de miR-17 al ARNm de PKD1 con un fármaco anti-miR-17 después de la formación de quistes también disminuyó el crecimiento de quistes, lo que indica que esta interacción podría ser un objetivo prometedor para el tratamiento de la enfermedad renal poliquística (PKD).
«Existen numerosas condiciones genéticas en las que una copia del gen causante está mutada, pero la otra copia sigue siendo normal. Nuestro enfoque para aprovechar la copia normal restante probablemente sea aplicable a muchas otras enfermedades además de la PKD», dijo el Dr. Patel.
Los resultados del estudio desafían el pensamiento actual sobre la poliquistosis renal autosómica dominante
Ronak Lakhia et al, PKD1 y PKD2 mRNA cis-inhibición impulsa la progresión de la enfermedad renal poliquística, Comunicaciones de la naturaleza (2022). DOI: 10.1038/s41467-022-32543-2
Citación: Los investigadores identifican un objetivo de terapia génica para la enfermedad renal poliquística (16 de septiembre de 2022) consultado el 16 de septiembre de 2022 de https://medicalxpress.com/news/2022-09-gene-therapy-polycystic-kidney-disease.html
Este documento está sujeto a derechos de autor. Aparte de cualquier trato justo con fines de estudio o investigación privados, ninguna parte puede reproducirse sin el permiso por escrito. El contenido se proporciona únicamente con fines informativos.


