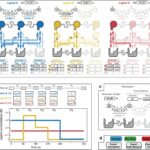
La interfaz de diseño de entrada de MVsim proporciona especificación de parámetros interactivos para sistemas de interacción multimolecular multivalente. a Una interfaz de apuntar y hacer clic permite al usuario seleccionar el número de ligandos (hasta tres) y las valencias de los ligandos y receptores (hasta trivalentes) que componen el sistema multivalente. Según el diseño elegido, el usuario especifica la estructura de cada uno de los ligandos ingresando el peso molecular aplicable (MW); los diámetros del dominio de unión (Ø); las longitudes de contorno (lc de los enlazadores (es decir, la distancia máxima de extremo a extremo; por ejemplo, 3,5 Å y 1,5 Å por aminoácido para una espiral aleatoria y una hélice alfa, respectivamente); y las longitudes de persistencia (lp) de los Además, se resaltan las interacciones combinatorias aplicables (numeradas del 1 al 9) únicas para cada emparejamiento de receptor-ligando. Los campos de parámetros permiten la entrada de constantes de velocidad monovalentes para cada interacción por pares. Las interacciones de no unión se pueden indicar con ken y kapagado valores de cero (p. ej., como se ilustra con el Ligando B en amarillo para las interacciones «1» y «5»). b Un campo de entrada permite al usuario especificar patrones de las concentraciones totales de ligandos a granel. Se produce una fase de asociación durante períodos de concentración de ligando a granel distinta de cero (p. ej., 90–270 s para el ligando B). Las fases de disociación ocurren cuando el ligando se elimina de la solución a granel (p. ej., 360 a 720 s para el ligando A). Aquí, el Ligando C se especifica como continuamente presente en solución durante los 720 s del transcurso del tiempo de interacción. La pantalla gráfica permite la visualización del patrón de pulso de concentración a granel especificado. C Parámetros de entrada de usuario para el receptor. La concentración del receptor se puede especificar como una densidad de superficie que imita la SPR (medida en RU; donde 1 RU equivale a ~1 pg/mm2) o una concentración molar. La topología del receptor se especifica de la misma forma que se ha descrito anteriormente para los ligandos. d los MV Sim La pestaña del controlador permite iniciar, iterar y exportar simulaciones vinculantes. «Iniciar» ejecuta una simulación. «Re-run» ejecuta una simulación abreviada utilizada cuando no se realizaron cambios en la valencia o la topología del sistema. «Restablecer» reinicia la aplicación y borra los parámetros de entrada del usuario de todos los campos. Crédito: Comunicaciones de la naturaleza (2022). DOI: 10.1038/s41467-022-32496-6
Un equipo dirigido por ingenieros biomédicos de la Universidad de Minnesota Twin Cities ha desarrollado una aplicación de acceso universal que puede simular interacciones moleculares complejas, lo que permitirá a los investigadores diseñar mejores tratamientos para enfermedades como el cáncer y la COVID-19.
El documento se basa en un estudio que los investigadores publicaron en 2019. Ahora, ampliaron la tecnología para simular interacciones moleculares aún más complejas, facilitaron el uso de la aplicación para los no expertos y aplicaron sus hallazgos para arrojar luz sobre cómo el SARS -El virus CoV-2 infecta el cuerpo.
El estudio se publica en Comunicaciones de la naturalezay la aplicación, llamada MVsim, está disponible gratuitamente para otros investigadores en GitHub.
El simulador predice la fuerza, la velocidad y la selectividad de las interacciones multivalentes, que involucran moléculas que tienen múltiples sitios de unión y pueden usarse para desarrollar medicamentos para enfermedades, particularmente el cáncer y el COVID-19.
«Las interacciones multivalentes son realmente importantes en los sistemas biológicos naturales, y ahora están comenzando a explotarse creativamente para crear nuevos medicamentos terapéuticos que aprovechen sus propiedades de unión únicas», dijo Casim Sarkar, autor principal del artículo y profesor de la Universidad de Minnesota. Departamento de Ingeniería Biomédica.
«Con los medicamentos multivalentes, en principio, puede atacar las células de manera muy específica de una manera que no es posible con los medicamentos monovalentes estándar, pero hay muchas variables a considerar en su diseño y hasta la fecha se ha realizado gran parte del trabajo en el campo. a través de ensayo y error experimental», agregó Sarkar. «Ahora, usando MVsim, podemos hacer buenas predicciones que pueden usarse para diseñar de manera más racional tales terapias».
Muchos medicamentos contra el cáncer no solo se unen a las células tumorales, sino también a las células a las que no están destinados, lo que a menudo crea efectos secundarios no deseados para el paciente. Al optimizar la especificidad de las interacciones multivalentes utilizando MVsim, los investigadores pueden diseñar fármacos que se dirijan más específicamente a las células de un tumor y minimicen la unión a otras células del cuerpo.
Otro ejemplo es el virus SARS-CoV-2. Los científicos saben que el virus está evolucionando para infectar mejor nuestras células y evadir nuestro sistema inmunológico, pero los mecanismos moleculares detrás de cómo el virus hace esto son relativamente desconocidos. Usando su tecnología MVsim, los investigadores de la Universidad de Minnesota pudieron explorar este proceso más a fondo, descubriendo las tasas a las que los dominios de unión individuales dentro de la proteína de pico multivalente del virus cambian entre un estado de infección celular y un estado de evasión inmune.
«Básicamente, tenemos un microscopio computacional que nos permite mirar debajo del capó y ver qué hacen las proteínas multivalentes, como la proteína espiga del SARS-CoV-2, a nivel molecular», explicó Sarkar. «Este nivel de detalle molecular es difícil de capturar con un experimento físico. Uno de los verdaderos poderes de MVsim es que no solo podemos aprender más sobre cómo funcionan estos sistemas, sino que también podemos usar esta herramienta para diseñar nuevas interacciones multivalentes para enfermedades como cáncer y COVID-19».
Los investigadores ya han identificado formas potenciales de limitar la infectividad de las variantes actuales y futuras del SARS-CoV-2, que planean probar pronto.
Proteína aglutinante autoensamblada multivalente diseñada contra SARS-CoV-2 RBD
Bence Bruncsics et al, MVsim es un conjunto de herramientas para cuantificar y diseñar interacciones multivalentes, Comunicaciones de la naturaleza (2022). DOI: 10.1038/s41467-022-32496-6
Aplicación MVsim: GitHub
Citación: La tecnología que simula interacciones moleculares complejas podría conducir a mejores tratamientos para el cáncer y la COVID-19 (6 de septiembre de 2022) consultado el 7 de septiembre de 2022 en https://phys.org/news/2022-09-technology-simulates-complex-molecular -interacciones.html
Este documento está sujeto a derechos de autor. Aparte de cualquier trato justo con fines de estudio o investigación privados, ninguna parte puede reproducirse sin el permiso por escrito. El contenido se proporciona únicamente con fines informativos.











