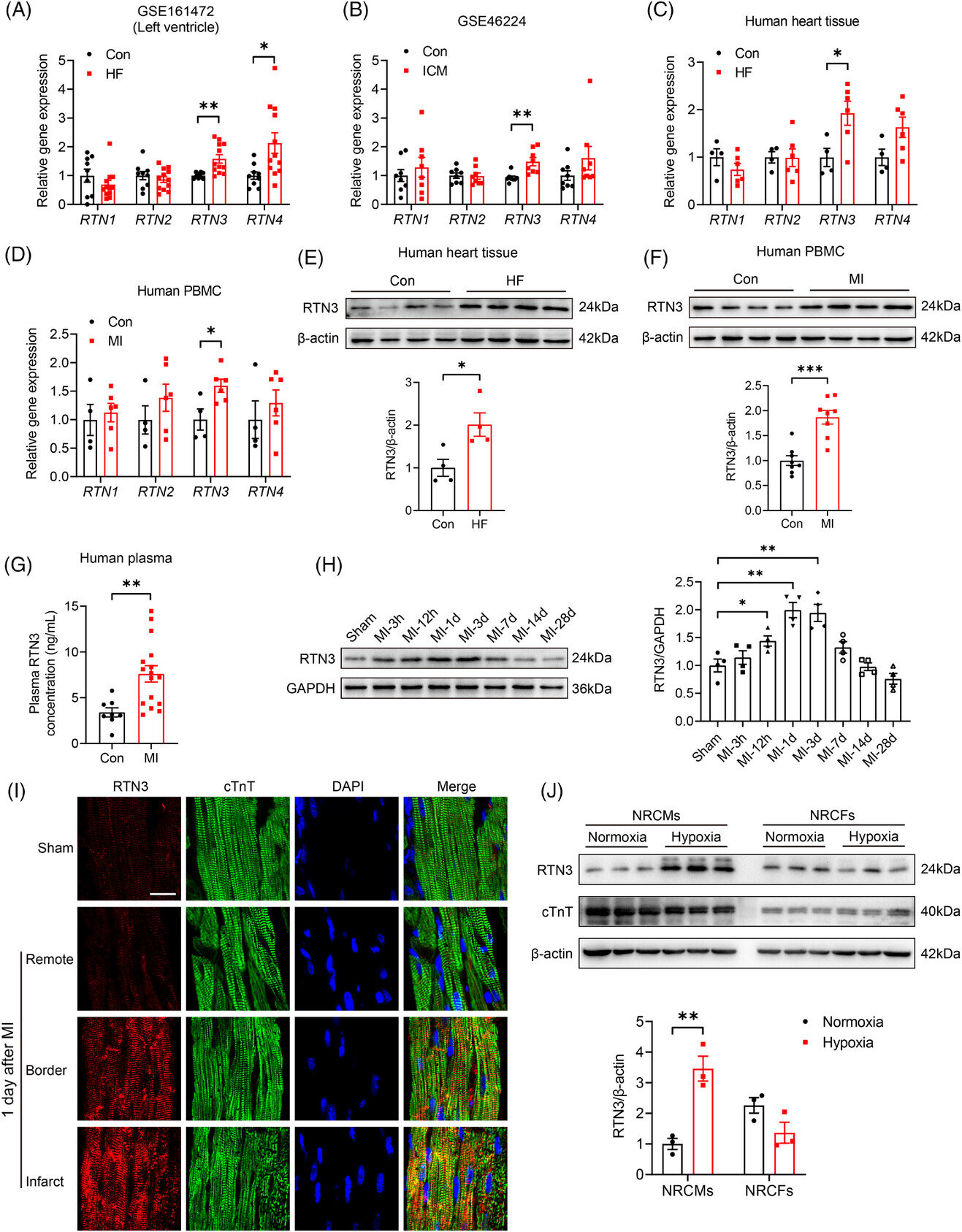
La expresión de reticulón 3 (RTN3) aumenta en el miocardio de pacientes con insuficiencia cardíaca (IC) y ratones con infarto de miocardio (IM). (A) Expresión transcripcional de RTN1, RTN2, RTN3 y RTN4. Crédito: MedCom (2024). DOI: 10.1002/mco2.503
La insuficiencia cardíaca (IC) después de un infarto de miocardio (IM) es un problema de salud mundial con una alta tasa de mortalidad. Múltiples mecanismos moleculares están implicados en el desarrollo de la insuficiencia cardíaca después de un infarto de miocardio. Sin embargo, las intervenciones dirigidas únicamente a estos procesos patológicos siguen siendo clínicamente ineficaces.
El Dr. Yan Li y el Dr. Mingming Zhang (Departamento de Cardiología, Hospital Tangdu, Universidad Médica de la Fuerza Aérea) han dirigido un estudio sobre este tema. El equipo descubrió que el reticulón 3 (RTN3), ubicado en el retículo endoplásmico (RE), está regulado negativamente en el tejido ventricular izquierdo de pacientes con insuficiencia cardíaca.
Los hallazgos son publicado en el diario MedCom.
Los investigadores construyeron un modelo de ratón con IM para investigar más a fondo las especificidades temporales y espaciales de la expresión de RTN3 en el miocardio después de un IM. Los resultados muestran que el aumento inducido por MI en el nivel de RTN3 se produce principalmente en la región del infarto y en la zona fronteriza del infarto en lugar de en la zona remota.
Luego, el equipo utilizó enfoques de ganancia y pérdida de función para investigar el papel de RTN3 en la insuficiencia cardíaca después de un infarto de miocardio. La sobreexpresión de RTN3 específica de cardiomiocitos disminuyó la función sistólica en ratones en condiciones fisiológicas y exacerbó el desarrollo de insuficiencia cardíaca inducida por IM. Por el contrario, la desactivación de RTN3 alivió la disfunción cardíaca después de un infarto de miocardio.
«Este resultado sugiere que RTN3 puede ser un objetivo terapéutico potencial para la insuficiencia cardíaca después de un infarto de miocardio», afirma el primer autor del artículo, Bingchao Qi.
Para explorar el mecanismo por el cual la desactivación de RTN3 mejora la insuficiencia cardíaca posterior a un infarto de miocardio, el equipo realizó co-inmunoprecipitación (Co-IP) y espectrometría de masas. A continuación, el ensayo Co-IP se realizó respectivamente utilizando el anticuerpo Flag y el anticuerpo HSPB1, confirmando que RTN3 interactuaba directamente con HSPB1.
Además, la eliminación de RTN3 redujo significativamente la expresión de HSPB1 en el RE y aumentó el contenido en el citoplasma, mientras que la sobreexpresión de RTN3 produjo el efecto opuesto. Como se esperaba, la eliminación de RTN3 disminuyó la expresión de TLR4 y aumentó la expresión de PGC-1α en el miocardio después del infarto de miocardio, mientras que la regulación positiva de RTN3 mediada por AAV9 aumentó significativamente la expresión de TLR4 y disminuyó la expresión de PGC-1α.
TLR4 puede inducir respuestas inflamatorias después de una lesión tisular. La inducción quirúrgica del IM dio como resultado una elevación de la expresión de TLR4 y del nivel de p-IκBα/IκBα, mientras que la eliminación de RTN3 revirtió las señales inflamatorias. La activación de la vía TLR4/p-IκBα es esencial para la fosforilación de NFκB y su translocación al núcleo.
Para verificar esto, el equipo extrajo los componentes nucleares de los tejidos cardíacos y demostró que la desactivación de RTN3 reducía la activación de NFκB y la translocación nuclear hasta cierto punto. El ensayo de inmunofluorescencia reveló una disminución significativa en la translocación nuclear de NFκB inducida por MI en los corazones de RTN3CKO ratones.
Además, PGC-1α es bien conocido como el regulador clave de la biogénesis mitocondrial. Por tanto, el equipo determinó el efecto de RTN3 en genes críticos de los complejos ETC mitocondriales. La regulación negativa de RTN3 aumentó los niveles de proteína de los genes relacionados con el complejo mitocondrial después de un infarto de miocardio; sin embargo, su regulación positiva ejerció un efecto contrario.
El suministro de energía juega un papel esencial en la cadena de transporte de electrones mitocondrial (CTE). Los investigadores observaron que la producción de ATP aumentaba en los corazones de ratones knockout para RTN3 y disminuía en los de ratones que sobreexpresaban RTN3.
Debido al papel mediador de HSPB1 en la disfunción cardíaca impulsada por RTN3, el equipo investigó si la inhibición farmacológica de HSPB1 podría contrarrestar los efectos protectores de la desactivación de RTN3 en la insuficiencia cardíaca después de un infarto de miocardio. Como se esperaba, la inhibición de HSPB1 inhibió significativamente el efecto protector de la desactivación de RTN3 sobre la función sistólica cardíaca en ratones después de un infarto de miocardio.
«La disfunción mitocondrial y la inflamación son patogénesis importante en varias enfermedades cardiovasculares, incluido el infarto de miocardio y la aterosclerosis. Por lo tanto, atacar RTN3 utilizando virus adenoasociados o inhibidores de moléculas pequeñas puede ser una estrategia potencial para prevenir y tratar la insuficiencia cardíaca posterior a un infarto de miocardio y otras enfermedades cardiovasculares. «, dice Qi.
Más información:
Bingchao Qi et al, La deficiencia de reticulón 3 mejora la insuficiencia cardíaca post-infarto de miocardio al aliviar la disfunción mitocondrial y la inflamación, MedCom (2024). DOI: 10.1002/mco2.503
Proporcionado por la Asociación de Promoción e Intercambio Médico Internacional de Sichuan
Citación: La deficiencia de reticulon 3 mejora la insuficiencia cardíaca posinfarto de miocardio al aliviar la disfunción mitocondrial (2024, 18 de abril) obtenido el 19 de abril de 2024 de https://medicalxpress.com/news/2024-04-reticulon-deficiency-ameliorates-myocardial-infarction. HTML
Este documento está sujeto a derechos de autor. Aparte de cualquier trato justo con fines de estudio o investigación privados, ninguna parte puede reproducirse sin el permiso por escrito. El contenido se proporciona únicamente con fines informativos.

