Crédito: Investigación de ácidos nucleicos (2025). Doi: 10.1093/nar/gkaf074
El síndrome de Rett es una enfermedad genética rara que afecta a 1 de cada 10,000 niñas recién nacidas, caracterizadas por una regresión repentina alrededor del 1 año de edad, con pérdida de lenguaje adquirido y habilidades motoras y conduciendo a un profundo deterioro cognitivo. Su causa principal son las mutaciones del gen MECP2, un controlador importante del desarrollo neuronal en el cerebro.
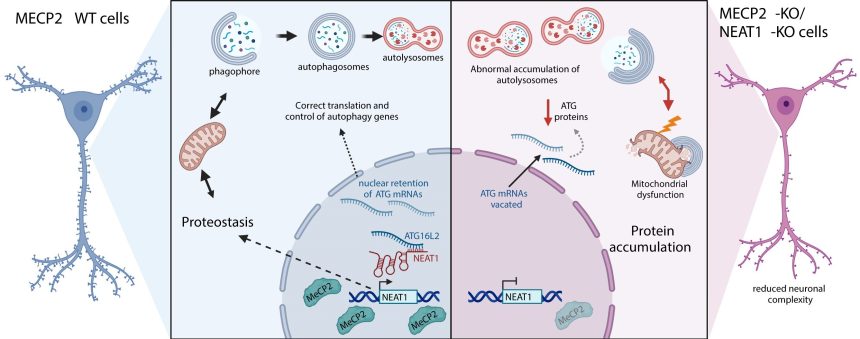
Precisamente para ser un controlador maestro, ha sido difícil descifrar cómo exactamente la pérdida de función de MECP2 conduce a las muchas alteraciones observadas dentro de las células cerebrales afectadas, como las anomalías morfológicas y los desequilibrios sinápticos. Una nueva investigación del laboratorio regulatorio de ARN y cromatina en el Instituto Josep Carreras arroja luz sobre una de estas alteraciones: el mal funcionamiento de la autofagia.
La vida interna de una célula es muy dinámica y necesita deshacerse de los componentes obsoletos con bastante frecuencia: ya no se necesitan proteínas, orgánulos que se expandieron para una tarea específica, etc. Uno de los sistemas que la célula usa para cumplir esta tarea es la autofagia, Literalmente «comer uno mismo», y es una parte importante del mantenimiento de la célula. Los defectos en el sistema de autofagia conducen a la acumulación de agregados de proteínas y otras deficiencias dentro de la célula, lo que resulta en una función anormal y consecuencias potencialmente graves.
En la investigación, primero autorizado por la Dra. Edilene Siqueira y recientemente publicado en el diario Investigación de ácidos nucleicosel equipo utilizó las últimas herramientas genéticas disponibles, como la secuenciación de ARN de una sola célula y la tecnología de edición de genes CRISPR/Cas9, y descubrieron que las mutaciones en MECP2 condujeron al agotamiento de un ARN largo no codificante llamado Neat1.
Como resultado, Neat1 controla el sistema de autofagia al establecer contactos directos de ARN-ARN con componentes de su maquinaria y dirigiendo su localización dentro de la celda. En consecuencia, la deficiencia de Neat1 contribuye a algunas de las alteraciones celulares encontradas en el síndrome de Rett.
Además, el equipo demostró que la restauración de Neat1 podría revertir estas alteraciones en los modelos in vitro de la enfermedad, abriendo la puerta a la exploración de nuevos enfoques terapéuticos en los próximos años.
La investigación es un esfuerzo conjunto entre el ARN regulatorio y el grupo de cromatina encabezado por el Dr. Sònia Guil, investigadores del Laboratorio de Epigenética del Cáncer dirigido por el Dr. Manel Esteller, también en el Instituto Josep Carreras, el Bellvitge Biomedical Research Institute (Idibell) y el Hospital Sant Joan de Déu.
Más información:
Edilene Siqueira et al, regulación mediada por Neat1 de proteostasis y localización de ARNm impacta la desregulación de la autofagia en el síndrome de Rett, Investigación de ácidos nucleicos (2025). Doi: 10.1093/nar/gkaf074
Citación: Estudio de mal funcionamiento de la autofagia se acerca un paso más para comprender el síndrome de Rett (2025, 20 de febrero) Recuperado el 20 de febrero de 2025 de https://medicalxpress.com/news/2025-02-autofagy-malfunction-closer-rett-syndrome.html
Este documento está sujeto a derechos de autor. Además de cualquier trato justo con el propósito de estudio o investigación privada, no se puede reproducir ninguna parte sin el permiso por escrito. El contenido se proporciona solo para fines de información.

