Gráficamente abstracto. Crédito: El Diario Americano de Genética Humana (2022). DOI: 10.1016/j.ajhg.2022.03.005
Mediante el uso de nuevos métodos para analizar los datos de ADN y los registros médicos, los investigadores de la Universidad de Brown están ayudando a mejorar la comprensión de los rasgos complejos que harán más descubrimientos relevantes para los grupos de ascendencia no blanca y no europea.
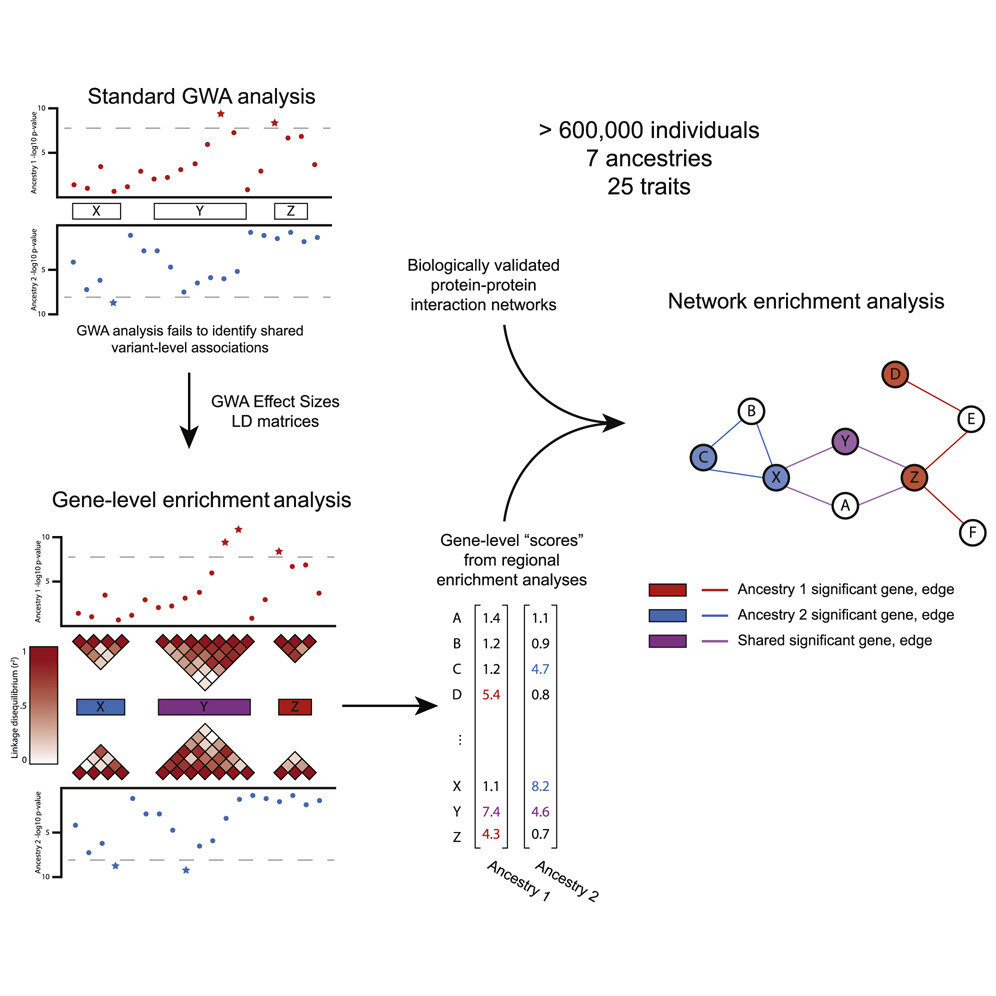
En un estudio publicado en la edición del 5 de mayo de El Diario Americano de Genética Humana, los investigadores ilustraron ejemplos de asociaciones robustas de determinantes de rasgos, o patrones de similitud, mientras estudiaban 25 rasgos en más de 600 000 individuos de siete ancestros humanos diversos. Con estas similitudes, los descubrimientos sobre la naturaleza de las enfermedades y sus respuestas a los tratamientos potenciales se vuelven más relevantes para grupos más grandes de personas, incluidas las poblaciones que anteriormente habían sido ignoradas o poco estudiadas.
Los conjuntos de datos de asociación de todo el genoma (GWA), que suelen utilizar los genetistas, se basan en la suposición de que las mutaciones genéticas individuales sustentan la base genética de los rasgos, explicó la autora del estudio Sohini Ramachandran, profesora de biología e informática que dirige el Centro para Biología Molecular Computacional y la Iniciativa de Ciencia de Datos en Brown. La idea es que un descubrimiento sobre esas mutaciones sea relevante para todas las personas en una variedad de grupos ancestrales diversos, de modo que si el hallazgo se usa para desarrollar tratamientos para afecciones genéticas, será aplicable para todas las personas con esa condición.
Sin embargo, estudios recientes han demostrado que los resultados de GWA estimados de personas europeas autoidentificadas no son transferibles a personas no europeas. Debido a esto, los conocimientos de los conjuntos de datos están sesgados en gran medida hacia el muestreo de individuos con ascendencia europea. La hipótesis estadística que subyace al marco GWA es injustamente restrictiva, dijo Ramachandran.
Por lo tanto, los investigadores utilizaron una nueva metodología de «análisis de enriquecimiento», desarrollada previamente en una colaboración entre Ramachandran y la profesora asistente de bioestadística de Brown, Lorin Crawford, para abordar el sesgo y la representación insuficiente.
«En este documento, que involucró un análisis muy cuidadoso de una gran cantidad de datos en múltiples biobancos, mostramos que los datos vistos solo a través de una lente GWA muy específica pueden parecer dispares e irreconciliables», dijo Ramachandran. «Sin embargo, visto de una manera más equitativa, con una metodología más expansiva, se vuelve biológicamente unificado, interpretable y, lo que es más importante, procesable».
El interés de Ramachandran en el tema comenzó cuando se enteró de un estudio que mostraba que los niños con leucemia linfoblástica aguda tenían diferentes respuestas al régimen de tratamiento estándar según su grupo de ascendencia; por ejemplo, los niños no blancos tenían más probabilidades de recaer y tenían un peor pronóstico. . Como biólogo evolutivo y genetista de poblaciones, Ramachandran comenzó a pensar en la creciente dependencia de los estudios GWA en el desarrollo de tratamientos «personalizados» para enfermedades y afecciones.
«No hubo mucha discusión sobre hasta qué punto los resultados de estos estudios iban a ser directamente aplicables a todos los ancestros», dijo. «Y según la teoría y la genética de poblaciones, parecía muy poco probable que esto fuera a funcionar de una manera equitativa».
Una nueva mirada a los datos
Con otros investigadores de Brown, incluidos Crawford y Samuel Pattillo Smith, Ramachandran comenzó a trabajar en el desarrollo de técnicas estadísticas que iban más allá de las mutaciones individuales para incluir genes y vías.
No es como si esta información no existiera; Durante las últimas dos décadas, las agencias de financiación y los biobancos de todo el mundo han realizado enormes inversiones para generar conjuntos de datos a gran escala de genotipos, exomas y secuencias de genomas completos de diversos ancestros humanos, que luego se fusionan con registros médicos y mediciones cuantitativas de rasgos. Sin embargo, explicaron los investigadores, los análisis de dichos conjuntos de datos generalmente se limitan a los análisis de asociación GWA que asumen una correlación directa entre mutaciones y rasgos.
Los investigadores estudiaron 25 rasgos en 566 786 personas de siete ancestros humanos autoidentificados diversos en el Biobanco del Reino Unido y el Biobanco de Japón, así como en 44 348 personas del Consorcio PAGE, incluidas cohortes de afroamericanos, hispanos y latinoamericanos, nativos hawaianos y estadounidenses. Individuos indios/nativos de Alaska. Realizaron pruebas estadísticas de asociación a nivel de mutación, gen y vía para los 25 rasgos cuantitativos.
Identificaron 1,000 asociaciones a nivel de genes que son significativas en todo el genoma en al menos dos grupos de ascendencia en estos 25 rasgos, así como asociaciones de vías en grupos europeos, asiáticos orientales y nativos de Hawái. La mayoría de estos no se habrían identificado usando GWA solo, dijeron los investigadores.
«En lugar de centrarnos en las pruebas estadísticas de mutación única, GWA, básicamente estamos abriendo un séquito más grande de pruebas que pueden buscar patrones a nivel genético o a nivel de vía anotada biológicamente», dijo Pattillo Smith, un Ph.D en biología computacional. . candidato en el laboratorio de Ramachandran. «Durante mucho tiempo, los científicos se han centrado tanto en el efecto de las mutaciones individuales que se ignora mucha información valiosa en los estudios de GWA o no se informa en las publicaciones resultantes, especialmente en las poblaciones de ascendencia que tienen cohortes más pequeñas porque la prueba en la mutación El nivel es increíblemente sensible a una serie de factores de confusión. Uno de los beneficios de agregar mutaciones al nivel de una región o gen es que puede suavizar esas cosas y ser más sólido en su detección del genoma a relación de rasgos».
Los investigadores apuntaban a lo que se llama «interpretabilidad biológica», dijo Ramachandran, «que es cómo podríamos implementar estos métodos para analizar los biobancos en toda su extensión y aprovechar toda la información que tienen para ofrecer».
Aplicación de metodología imparcial a conjuntos de datos sesgados
En el artículo, los investigadores discuten cómo los biobancos están fuertemente sesgados hacia las personas que se identifican a sí mismas como de ascendencia europea, señaló Crawford, profesor asistente de bioestadística en Brown afiliado al Centro de Biología Molecular Computacional. Una joya escondida de esta nueva investigación, dijo Crawford, es que muestra cómo el desarrollo de métodos estadísticos sofisticados puede ayudar a superar limitaciones como una muestra subrepresentada de grupos de ascendencia no europea.
«No tienes que esperar hasta que la cantidad de personas de otros grupos de ascendencia sea igual a la cantidad de personas que se identifican a sí mismas como europeas», dijo Crawford. «De hecho, incluso si se generan más datos, es probable que se perpetúe el mismo desequilibrio. Mientras tanto, los métodos estadísticos a escalas más altas de genes y vías aún pueden ayudarnos a obtener información sobre la arquitectura genética que se puede aplicar de manera beneficiosa». a estos grupos de ascendencia subrepresentados. Esta metodología puede ayudarnos a usar los datos de manera más equitativa, en este momento».
En un campo como la genómica, hay mucho en juego, dijo Ramachandran.
«Es muy importante para nosotros que entendamos mejor la arquitectura de rasgos para que podamos dar pasos para proporcionar terapias efectivas para todos, de cada grupo de ascendencia».
Las puntuaciones de riesgo poligénico identifican a las personas de alto riesgo en ascendencia europea y asiática, pero menos en ascendencia africana
Samuel Pattillo Smith et al, Los análisis de enriquecimiento identifican asociaciones compartidas para 25 rasgos cuantitativos en más de 600,000 individuos de siete ancestros diversos, El Diario Americano de Genética Humana (2022). DOI: 10.1016/j.ajhg.2022.03.005
Citación: Los investigadores destacan una forma más equitativa de analizar los datos de ADN de grupos no estudiados (4 de mayo de 2022) consultado el 5 de mayo de 2022 en https://medicalxpress.com/news/2022-05-highlight-equitable-dna-understudied-groups.html
Este documento está sujeto a derechos de autor. Aparte de cualquier trato justo con fines de estudio o investigación privados, ninguna parte puede reproducirse sin el permiso por escrito. El contenido se proporciona únicamente con fines informativos.


